



- Pharmaceuticals
- Pharmaceutical Regulatory Affairs Market
Pharmaceutical Regulatory Affairs Market Size, Share, and Growth Forecast, 2025 - 2032
Pharmaceutical Regulatory Affairs Market By Service Type (Regulatory Consulting, Legal Representation, Others), Product (Drugs, Biologics, Medical Devices), Development Stage (Preclinical, Clinical Trials, Post-Approval/Post Market Authorization (PMA)), and Regional Analysis for 2025 - 2032
Pharmaceutical Regulatory Affairs Market Share and Trends Analysis
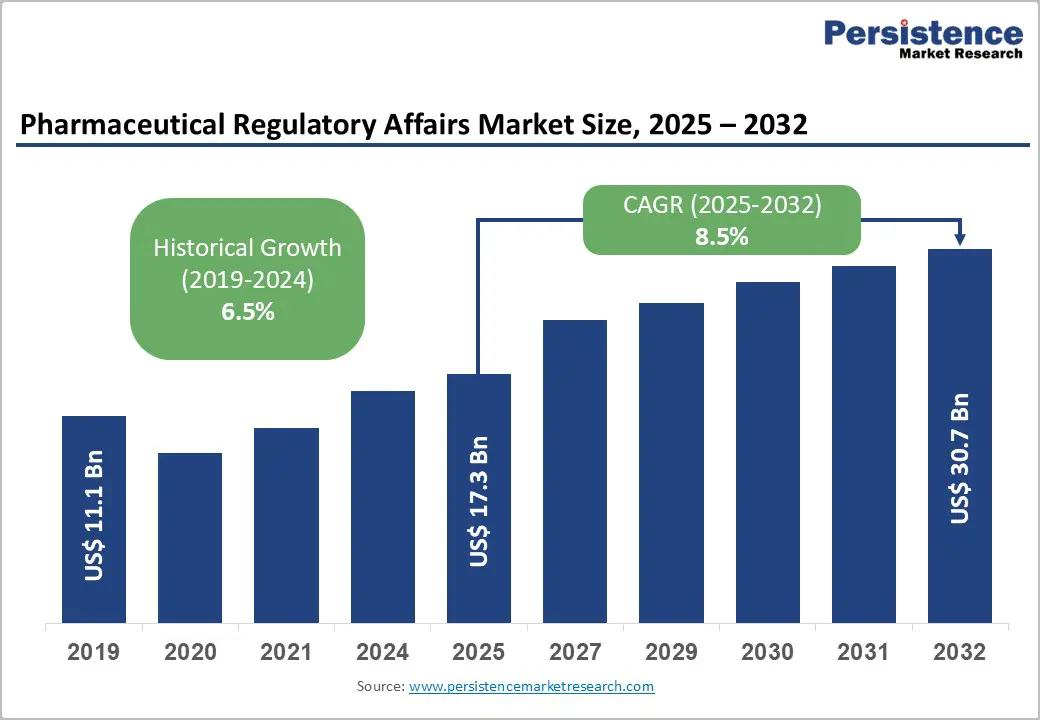
The global pharmaceutical regulatory affairs market size is likely to be valued at US$17.3 Billion in 2025, and is estimated to reach US$30.7 Billion by 2032, growing at a CAGR of 8.5% during the forecast period from 2025 to 2032, driven by increasing complexity in drug approval pathways, the rapid expansion of biologics and advanced therapy medicinal products (ATMPs), and regulatory harmonization efforts across major markets.
Market growth is driven by pharmaceutical and biotech companies increasingly outsourcing regulatory functions to manage evolving compliance demands, accelerate approvals, and reduce operational costs. Innovations such as the FDA’s Breakthrough Therapy Designation and the EMA’s PRIME program further boost the need for specialized regulatory expertise.
Key Industry Highlights
- Product Leadership: Biologics represent the fastest-growing segment, driven by gene therapy and cell therapy development requiring specialized regulatory submissions.
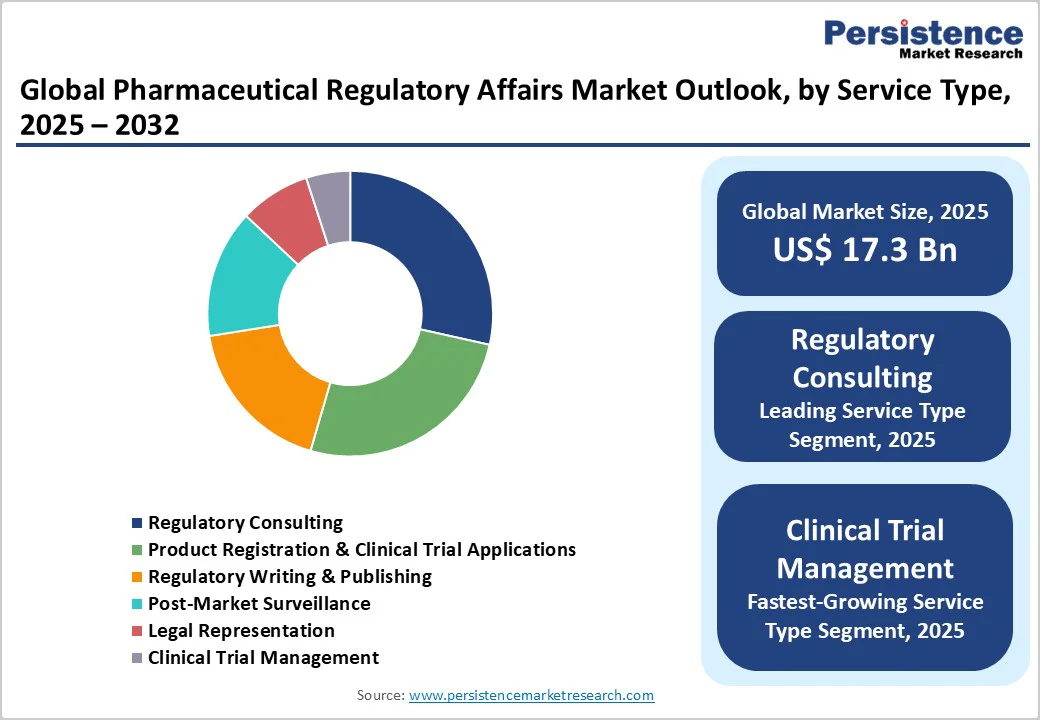
- Fastest-growing Service Type: Clinical trial management services constitute the fastest-growing service segment from 2025 to 2032, propelled by proliferating complex clinical trials requiring sophisticated regulatory oversight.
- Leading Service Type: Regulatory consulting maintains market leadership at 28.5% in 2025, reflecting an urgent need for strategic guidance in navigating multi-jurisdictional frameworks.
- Dominant Region: Asia Pacific dominates the global market with about 37.5% share in 2025 and leads growth at the highest CAGR through 2032, driven by clinical trial proliferation and regulatory modernization in China and India.
- Regulatory Landscape: Breakthrough Therapy Designation reached 25% of FDA drug approvals between 2021 and 2023, creating substantial demand for regulatory expertise for navigating expedited pathways and compressed development timelines.
- Regional Scenario: The EMA approved a record 28 biosimilars in 2024, demonstrating regulatory momentum supporting biosimilar market expansion and generating sustained regulatory affairs service demand.
- October 2025: Parexel partnered with Weave Bio to accelerate regulatory submissions by integrating Weave’s AI-driven platform with Parexel’s regulatory consulting expertise, enabling faster and higher-quality Investigational New Drug (IND) applications and clinical trial initiations.
| Key Insights | Details |
|---|---|
| Pharmaceutical Regulatory Affairs Market Size (2025E) | US$17.3 Bn |
| Market Value Forecast (2032F) | US$30.7 Bn |
| Projected Growth (CAGR 2025 to 2032) | 8.5% |
| Historical Market Growth (CAGR 2019 to 2024) | 6.5% |

Market Factors - Growth, Barriers, and Opportunity Analysis
Proliferation of Accelerated Regulatory Pathways Transforming Drug Development
The market growth is driven by the widespread adoption of accelerated approval mechanisms that fundamentally alter the economics and timelines of drug development.
For example, between 2021 and 2023, the FDA granted Breakthrough Therapy Designation (BTD) to 25% of all approved drugs. Expedited pathways such as these are compressing development timelines by several months for qualifying therapies, particularly in oncology, rare diseases, and gene therapies.
With clinical trial delays costing pharmaceutical companies millions, they are investing heavily in regulatory affairs capabilities to accelerate submissions and reduce approval timelines. The 21st Century Cures Act in the U.S. has further streamlined FDA processes for breakthrough therapies, while the updated 2024 FDA draft guidance enables interchangeability designation for biosimilars.
These regulatory innovations are creating an explosive demand for consultants who can navigate complex submission requirements, prepare accurate documentation, and leverage AI-assisted submission management systems that are digitalizing regulatory workflows.
Escalating Compliance Costs and Regulatory Fragmentation Creating Barriers
Limitations on the pharmaceutical regulatory affairs space stem from escalating compliance costs and persistent regulatory fragmentation across jurisdictions. Global pharmaceutical companies spend billions annually on regulatory compliance, with individual significant compliance failures triggering massive remediation.
Smaller pharmaceutical and biotech companies are disproportionately affected, as regulatory compliance represents a substantial percentage of operational budgets for firms lacking scale economies. This cost burden creates formidable barriers to entry, particularly for innovative startups developing novel therapeutics that require extensive regulatory documentation and multi-jurisdictional approvals.
Regulatory harmonization remains incomplete despite International Council for Harmonisation (ICH) efforts. This fragmentation necessitates maintaining 3-4 versions of core quality documents to satisfy competing requirements, increasing the documentation maintenance burden significantly.
Cross-market product introductions face additional costs, going up to US$2.5 Million per product due to divergent regulatory expectations. This is particularly evident in advanced therapy products where the FDA and EMA maintain substantially different evidentiary standards for cell and gene therapies.
Advanced Therapy Medicinal Products and Biosimilars Creating High-Value Service Opportunities
The most lucrative opportunity within the market lies in specialized services for ATMPs, encompassing gene therapies, cell therapies, and tissue-engineered products, and the expanding biosimilars segment.
With each biosimilar requiring complex regulatory submissions to demonstrate comparability with reference biologics, the scope for regulatory specialists capable of navigating biologics-specific pathways, including the FDA's 351(k) pathway and EMA's biosimilar guidelines, is immense.
ATMPs represent an even more transformative opportunity, with regulatory affairs for biotech products tightening rapidly, significantly outpacing traditional small-molecule drugs.
Gene and cell therapies command premium regulatory service pricing due to their complexity, with specialized ATMP regulatory submissions requiring expertise in manufacturing controls, product characterization, and long-term patient monitoring protocols that differ drastically from conventional pharmaceuticals.
Canada's Biosimilars Initiative, now implemented in 12 of 13 provinces and territories, mandates patient transitions to approved biosimilars under public PharmaCare coverage, creating a relatively secure market for biosimilar developers and corresponding regulatory service demand.
Category-wise Analysis
Service Type Insights
Regulatory consulting dominates, capturing about 28.5% market revenue share in 2025, boosted by the fundamental complexity of navigating multi-jurisdictional regulatory frameworks where pharmaceutical companies require strategic guidance spanning submission strategy, regulatory pathway selection, and compliance program design.
Regulatory consultants provide high-value advisory services that extend beyond transactional document preparation to encompass strategic decision-making regarding optimal market entry sequences, expedited pathway eligibility assessment, and regulatory risk mitigation.
The segment benefits from regulatory landscape evolution, with continuous changes to FDA, EMA, NMPA, and other health authority requirements generating persistent demand for specialized expertise.
Clinical trial management services represent the fastest-expanding segment through 2032, attributable to the proliferation of complex clinical trials for ATMPs, gene therapies, and personalized medicine applications that require sophisticated regulatory oversight throughout the development lifecycle.
The segment encompasses clinical trial application preparation, protocol review and approval, ongoing trial monitoring for regulatory compliance, and management of interactions with health authorities during active investigations.
Rising clinical trial registrations globally, particularly in Asia Pacific, where the volume of trials is the highest worldwide, are driving the demand for specialized services that ensure adherence to Good Clinical Practice (GCP) standards and jurisdiction-specific requirements.
Product Category Insights
Traditional pharmaceutical drugs command the largest revenue share at 52% in 2025, reflecting the segment's historical dominance and continued volume leadership despite slower relative growth. These products benefit from established regulatory frameworks, mature approval pathways, and continuous pipeline replenishment as pharmaceutical companies develop next-generation versions of proven therapeutic classes.
Small-molecule drugs maintain regulatory affairs demand through lifecycle management activities, including indication expansions, formulation modifications, and geographic market extensions that each require comprehensive regulatory submissions.
Generic drug approvals, while following streamlined pathways such as the FDA's Abbreviated New Drug Application (ANDA) process, still generate substantial regulatory service demand given the volume of applications and bioequivalence documentation requirements.
Biologics constitute the fastest-growing product category from 2025 to 2032. Its exceptional growth is supported by the increasing pharmaceutical inclination toward large-molecule therapeutics offering targeted mechanisms of action and addressing previously untreatable conditions.
Biosimilars have been experiencing approval spikes from the FDA and EMA over the past decade, driven by patent expirations of blockbuster biologics, including Humira (adalimumab), Herceptin (trastuzumab), and emerging opportunities as additional reference products lose exclusivity through 2030. ATMPs represent the segment's most dynamic component, requiring specialized regulatory expertise far exceeding conventional drug submissions.
Development Stage Insights
The clinical trials development stage leads with a market share of approximately 46.5% in 2025, due to the concentration of regulatory activity during active clinical investigation when pharmaceutical companies face intensive health authority interactions, protocol modifications, and submission requirements.
This stage encompasses IND applications, Clinical Trial Applications (CTAs), protocol amendments, safety reporting, and ongoing communications with regulatory bodies throughout Phase I, II, and III studies. The segment's leadership position stems from the duration of clinical trial development, which typically spans multiple years, and the regulatory intensity required to satisfy evolving safety and efficacy evidence standards.
Post-approval regulatory affairs, encompassing post-market surveillance, pharmacovigilance, lifecycle management, and ongoing compliance activities, represents the fastest-growing segment through 2032. This accelerated growth reflects intensifying health authority scrutiny of marketed products, expanding pharmacovigilance requirements, and the cumulative effect of approved product portfolios requiring continuous regulatory maintenance.
Regulatory agencies globally are emphasizing post-market safety monitoring, with the FDA and EMA implementing enhanced surveillance systems, including Sentinel Initiative and EudraVigilance, which require pharmaceutical companies to submit comprehensive adverse event reports and conduct post-marketing studies.
Regional Insights
North America Pharmaceutical Regulatory Affairs Market Trends
North America remains the second-largest market, holding a 33% share in 2025. The U.S., as the innovation hub with the FDA’s rigorous regulatory framework and the highest pharmaceutical R&D investments, drives most of this activity, supported by biopharma clusters in Boston, San Francisco, and San Diego.
Canada adds value through biosimilar adoption and expanding clinical trials. The FDA’s sophisticated processes, including expedited pathways such as the BDT Designation, generate sustained demand for expert regulatory services, with enforcement penalties reinforcing compliance importance.
The competitive landscape features global contract research organizations (CROs) such as IQVIA and Parexel alongside niche consultancies, with FDA approval often serving as a global benchmark, facilitating international market access.
Growth is fueled by biologics and ATMP expansion, biosimilar maturation, and rising complexity from digital health and AI diagnostics. Despite a moderate CAGR due to market maturity and clinical trial shifts to Asia Pacific, North America continues to lead in strategic regulatory consulting and breakthrough innovation support, retaining its role as a global quality and innovation standard.
Europe Pharmaceutical Regulatory Affairs Market Trends
Europe is anticipated to grow at a steady CAGR through 2032. The EMA centralizes approvals for all 27 EU countries, supported by national pathways, with Germany, France, the U.K., Spain, and Italy driving about 70% of the activity. Post-Brexit, the U.K.’s Medicines and Healthcare products Regulatory Agency (MHRA) requires separate submissions, adding complexity.
The EMA’s rigorous framework parallels the FDA’s and offers expedited pathways such as PRIME and conditional marketing authorizations, while approving a record 28 biosimilars in 2024, underscoring Europe’s biosimilar leadership fueled by cost containment policies.
Europe’s specialized advanced therapy medicinal product (ATMP) regulation fosters gene and cell therapy development. The strong focus on pharmacovigilance, with EudraVigilance managing over 4 million adverse event reports annually, amplifies the demand for post-market services.
Growth is driven by personalized medicine, oncology expansion, and digital regulatory modernization, though demographics and maturity temper CAGR, positioning Europe as a vital, sophisticated global market actor.
Asia Pacific Pharmaceutical Regulatory Affairs Market Trends
Asia Pacific leads with an estimated 37.5% share in 2025 and the highest CAGR through 2032, making it the largest and fastest-growing regional market for pharmaceutical regulatory affairs. This growth is fueled by expanding clinical trial activities, rapid pharmaceutical manufacturing scale-up, regulatory modernization, and robust domestic innovation in China, Japan, India, and ASEAN countries.
China dominates regional growth, driven by reforms such as the 2015 Marketing Authorization Holder (MAH) system, which reduced drug approval timelines for priority products.
India’s role as the “pharmacy of the world” and its streamlined clinical trial regulations attract multinational sponsors, while Japan’s sophisticated Pharmaceuticals and Medical Devices Agency (PMDA) demands specialized regulatory expertise. ASEAN countries are progressively harmonizing regulations, though implementation remains uneven.
The regional market also benefits from lower clinical trial costs, large treatment-naïve populations, expanding contract manufacturing, and a skilled, cost-competitive workforce. Increasing R&D investments and outsourcing by pharmaceutical companies are reinforcing Asia Pacific’s rapid ascent, with projections to surpass 40% market share by 2032 amid continued regulatory evolution and innovation across diverse markets.

Competitive Landscape
The global pharmaceutical regulatory affairs market is moderately fragmented, with major CROs such as IQVIA, Parexel, and ICON holding about a 25% share due to their global networks and integrated services.
Mid-tier players such as Labcorp and Syneos add another 10-15%, while niche firms including Freyr Solutions, WuXi AppTec, and Charles River Laboratories serve specialized needs. Competition is driven by therapeutic expertise, regional capabilities, and tech adoption such as AI-enabled submission tools. Despite gradual consolidation, fragmentation remains due to diverse client requirements and demand for specialized regulatory knowledge.
Key Industry Developments
- In November 2025, the FDA finalized guidance on patient-focused drug development (PFDD), promoting the use of patient experience data in regulatory decisions. It encourages early patient engagement to inform benefit-risk assessments and clinical study design, ensuring therapies effectively meet patient needs and advance patient-centered innovation.
- In October 2025, Thermo Fisher Scientific announced a US$9.4 Billion acquisition of Clario, enhancing its clinical development and laboratory services portfolio. Clario’s expertise in trial management and data solutions complements Thermo Fisher’s capabilities, aiming to accelerate biopharmaceutical product development and strengthen competitiveness in the evolving life sciences services market.
- In September 2025, IQVIA launched the AI-powered Clinical Trial Financial Suite (CTFS) to streamline budgeting, contracting, forecasting, and payments. Its first module, CTFS Site Payments, automates site payment workflows, cutting processing time by up to 50%, offering scalable, secure, and efficient end-to-end financial management for sponsors, CROs, and study sites.
Companies Covered in Pharmaceutical Regulatory Affairs Market
- IQVIA Holdings Inc.
- Parexel International Corporation
- ICON plc
- Labcorp Drug Development
- Syneos Health, Inc.
- WuXi AppTec
- Charles River Laboratories International, Inc.
- PRA Health Sciences
- Medpace Holdings, Inc.
- Freyr Solutions
- Pharmalex GmbH
- Genpact Limited
- Pharmexon
- Criterium, Inc.
- Clarivate Analytics
Frequently Asked Questions
The global pharmaceutical regulatory affairs market is projected to reach US$17.3 Billion in 2025.
Escalating demand for specialized regulatory expertise driven by increasing complexity in drug approval pathways, the rapid expansion of biologics and ATMPs, and regulatory harmonization efforts across major markets are driving the pharmaceutical regulatory affairs market.
The pharmaceutical regulatory affairs market is poised to witness a CAGR of 8.5% from 2025 to 2032.
Rising outsourcing of regulatory functions by pharma and biotech firms, coupled with innovations such as the FDA’s Breakthrough Therapy Designation, drives market growth by streamlining compliance, speeding approvals, and reducing operational costs.
IQVIA Holdings Inc., Parexel International Corporation, and ICON plc are some of the key players in the pharmaceutical regulatory affairs market.





